Single cell proteomics is a field in rapid development and holds the potential to transform the landscape of cell biology, offering an unbiased glimpse into the proteome at the individual cell level. Despite its revolutionary possibilities, single cell proteomics is still in its early stages and is faced with challenges to scale the number of cells for analysis needed to gain biological insight from single cell proteomics. This study led by the Olsen group at University of Copenhagen introduces significant methodological advancements that significantly enhance the sensitivity, coverage and reliability of protein identification in single cells.
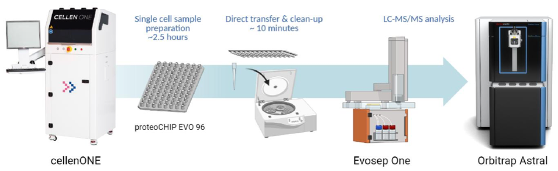
An important step towards scalable single cell proteomics is streamlined workflows with high sensitivity to minimize surface adsorption losses. This is enabled by the recently introduced proteoCHIP EVO 96 from Cellenion. This ensures precise isolation and digestion of individual cells in an automated manner. The format of the proteoCHIP is designed to enable pipetting free transfer of samples into Evotips, which serves as the sample introduction device for the Evosep One. Sensitive liquid chromatography is then enabled by the Whisper methods coupled to efficient narrow-window data independent acquisition (nDIA) on the Thermo Scientific Orbitrap Astral mass spectrometer.
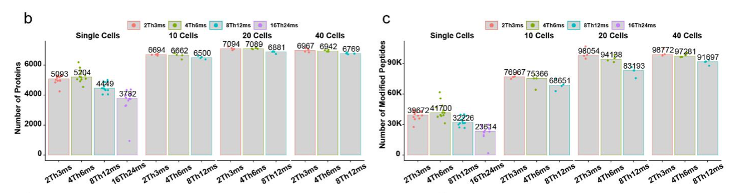
Through a comparative analysis, the optimal nDIA settings for single cells were found using 4 Th DIA windows and 6 ms max IT (4Th6ms). This resulted in the highest proteome coverage, leading to the identification of a median number of proteins of 5204 proteins with a significant depth of more than 40,000 modified peptides. For higher input materials of 20 cells, more than 7,000 proteins were identified from close to 100,000 modified peptides.
Direct analysis of PTMs from single cells without enrichment
The Orbitrap Astral mass spectrometer measures an exceptional depth of peptide precursors from low sample amounts and the authors found that this enables the identification of PTMs directly from single cells. As these are sub-stoichiometric, global analysis typically requires specific enrichment of the PTM bearing peptides prior to MS analysis, but with the high peptide coverage in the dataset, they were able to identify close to 170 phosphorylation sites highlighting prevalent phosphorylation motifs like the SP motif corresponding to substrates for abundant kinases like CDKs.
In conclusion, the workflow is the first commercially available workflow for scalable single cell analysis with the ability to isolate and digest 192 single cells at the time using the cellenONE and directly analyze the samples using high-throughput LC-MS.
It is set to be a cornerstone in future single cell proteomics studies due to the ease-of-use allowing us to analyze the number of single cells which are needed to get valuble biological insight and can be expanded to uncover the exciting potential in spatial proteomics, where input material is also limited.
Read more about the work here