Update: Check out our latest on single cell proteomics here.
Single-cell technologies are revolutionizing biology but have so far mainly been limited to imaging and deep sequencing. However, proteins are the main drivers of cellular function and in-depth characterization of individual cells by mass spectrometry (MS)- based proteomics would thus be highly valuable and complementary. A collaboration between Evosep, Bruker and the Helmholtz Zentrum in Münich, led by the Mann group at the Max Planck Institute for Biochemistry, has made this a reality.
The ultra-high single-cell sensitivity was achieved by innovative developments throughout the entire workflow, from a nearly loss-less miniaturized sample preparation workflow to a new ultra low-flow method on a standard Evosep One, delivered with a gradient flow of 100 nl/min, leading to approximately 10 times higher sensitivity than gradients at 1 µl/min, and finally a new modified timsTOF mass spectrometer with a brighter ion source. Combined, these innovations present a scalable, quantitative, and ultra-high sensitivity workflow dedicated to routinely measure cohorts of single cells, and ultimately reveal new cell biological information.
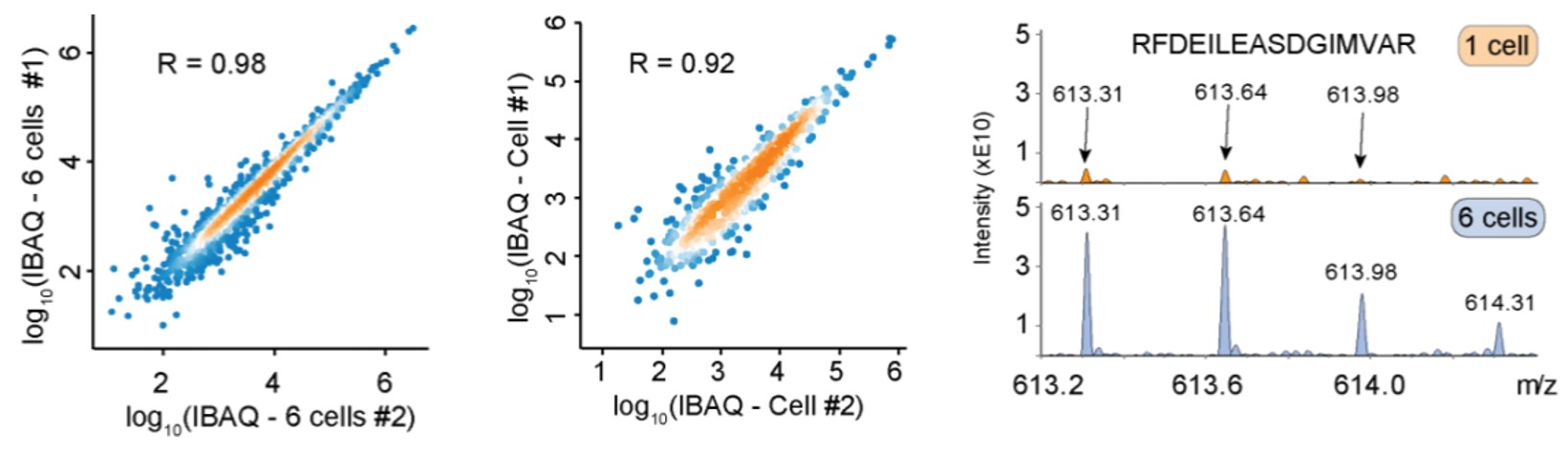
To illustrate the sensitivity, they FACS sorted 0, 1 and up to 6 HeLa cells in triplicate and processed them with the described workflow using ddaPASEF. Quantification accuracy was high when comparing single cells, not much reduced from comparing six cells. When they inspected the lowest abundant peptide that was shared between all samples, it showed that clearly interpretable fragment ion series were still present at high signal to noise levels.
To investigate if their proteomics workflow could detect biological responses to drug perturbation at the single-cell level, they treated HeLa cells with thymidine and nocodazole to produce four cell populations enriched in specific cell cycle stages. The analysis comprised a total of 221 single cells and they quantified up to 1,209 proteins per single cell and 1,305 overall using diaPASEF.
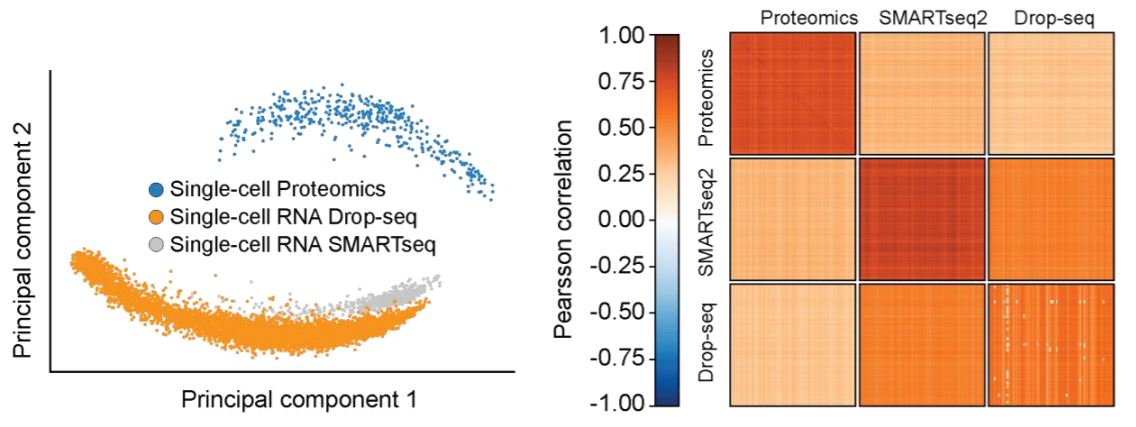
In addition to these drug-perturbed cells, they measured more than 200 untreated ones and compared the set of more than 420 single-cell proteomes to similar single-cell RNA sequencing data. Single-cell protein and RNA levels are very different, which implies distinct RNA and protein abundance regulation mechanisms. Protein measurements thus yield complementary quantitative biological information.
Results also implies that single cells have a stable core proteome whereas this is not the case for the transcriptome.
FUTURE DIRECTIONS
The work presented, demonstrates the sensitivity gain for single-cell total proteome measurements but will be advantageous in any situation that is sample limited. This includes investigation of post-translational modifications from small numbers of cells or from in vivo material, measurements directly from paraffin embedded formalin fixed (FFPE) pathology specimens, and the analysis of other compound classes such as metabolites or drugs.
WHISPER IS HERE
Watch our Whisper webinar from December 15th, where Andreas-David Brunner presents his data on single-cell proteomics.
→ Read full publication on embopress
→ Fore more data using Whisper 40SPD click here
LET’S STAY IN TOUCH
Join our monthly newsletter to stay tuned on new data and publications using Evosep One technology.