WHAT MATTERS IN CLINICAL PROTEOMICS? 1. CHALLENGES OF LARGE COHORT STUDIES
By Nicolai Bache
Cohort studies are essential in the discovery and validation of protein biomarkers. Every day, clinical proteomics labs evaluate patient samples to correlate protein patterns with a specific status – e.g., healthy vs. pathogenic, or therapeutically responsive vs. non-responsive.
The enormous amount of data required to analyze large sample cohorts calls for suitable methods. Liquid chromatography mass spectrometry (LC/MS) provides high precision, but precision and depth of analysis take time and limit throughput. Verification by LC/MS furthermore competes against other approaches. Certainly, proteomics cohort studies are very demanding projects, but what exactly does that mean?
- What are the pain points and needs of LC/MS proteomics labs?
- Who exactly is doing cohort studies, and what is the real scale of a “large” study?
- How long does a typical study take and why?
- How can we facilitate LC/MS–based cohort studies, the discovery and validation of biomarkers and, ultimately, improved patient care?
In a recent survey, conducted from November 2016 to February 2017, we asked exactly these questions. More than 70 scientists took the time to respond – group leaders, postdocs, students and engineers, working in academia, industry, and hospitals all over the world.
CHALLENGES IN PROTEOMICS COHORT STUDIES
More than one third of the survey respondents are doing large proteomics cohort studies, and another 26.7% are getting started, indicating high prevalence and significant growth. At the same time, the scale of these studies varies greatly. Conducting a study takes about nine days on average, ranging from hours to over 100 days. The time requirement per study depends on two factors: the number of samples, which can be from very few to 2000 samples (200 on average) and the time per measurement (less than a minute to 4 hours, with an average around 90 minutes).
However, the quantity of samples typically processed in a lab does not correlate with the measurement time per sample, so that we cannot distinguish typical features of high- or low-throughput labs. Even though some labs analyze thousands of samples per study, processing each sample can take up to several hours. Especially in these cases, a faster workflow could offer significant cost savings.
Another reason for the large variety in time per study is that labs use their LC/MS equipment for a wide range of different applications – from protein identification through biomarker discovery and validation to more specialized applications like human leukocyte antigen (HLA) typing. The time and effort needed to obtain the required results depends significantly on the questions asked in the particular study. When designing experiments for clinical studies, it is critical that the protocols can easily be repeated by other labs for verification.
Now to the challenges. We looked at the proteomics workflow to identify the most pressing bottlenecks (Fig.1). In a ranking of workflow steps, robustness in the LC step was regarded as most critical, especially for those users working in the hospital or clinic environment. Sample preparation was rated as second most important bottleneck, followed by MS robustness and data analysis. Sample availability however was not critical for most respondents.
Faster and more robust LC and sample preparation procedures could therefore improve the proteomics workflow for most users.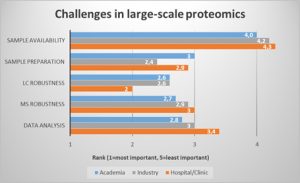
Fig. 1: LC robustness and sample preparation are the major challenges in the large-scale proteomics workflow.
(Question: Rank the following challenges related to large-scale proteomics studies. 1 (=most important) to 5 (least important). Values were averaged and filtered by respondents who indicated they worked in hospital/clinic, industry, or academic laboratories.
INCREASING THROUGHPUT
What about the needs in LC/MS based proteomics studies? Overall, data precision and reproducibility were regarded as most important. Not surprisingly, throughput was ranked higher by scientists working in the hospital/clinical environment than by those in industry or academia (Fig.2). This is reflected by the larger number of samples per study (219 on average in the hospital/clinic vs. 138 in academia). At the same time, almost half of the respondents across the different working environments are either paid by sample (31%) or by study (11.3%), so that an increase in throughput means a direct financial benefit to them.
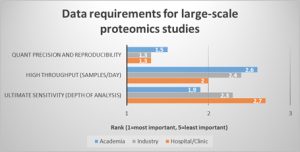
Fig. 2 Quantification precision and reproducibility is the most important data requirement across all working environments, whereas throughput is highly relevant in the hospital/clinic.
(Question: Rank your data requirements for large-scale proteomics studies. 1 (=most important) to 5 (least important). Values were averaged and sorted by respondents who indicated they worked in hospital/clinic, industry, or academic laboratories.
How can users process more samples per day? For most respondents (84.5%), simply buying more instruments is not an option. This is only partly due to financial constraints. Speeding up the LC step, such as by reducing the gradient, was the most frequently ticked answer (see Fig. 1). Therefore, optimizing the LC step is believed to be more effective in increasing throughput than investing in additional equipment.
Addressing the need for faster workflows and higher reproducibility in LC/MS–based cohort studies is what we do at Evosep. Our mission is to significantly improve the robustness and speed of sample preparation and sample separation. Listening to the needs of users in clinical proteomics helps us take the right steps to achieve this goal.
1 Comment
Submit a Comment
Your comment will be visible after moderation (usually within 24 hours).
Most proteomic investigations today are based on bottom-up approaches and are wrongly interpreted. Peptide data are interpreted wrongly as protein synthesis data. This basic concept is wrong. Not proteins, but protein species are the functional molecules within the proteome. Top-down proteomics will clearly better describe reality. The clinically relevant molecules are only reached at the protein species level. Prediction: The protein species concept will soon overcome the protein expression discourse. For references see: Jungblut et al. Chem Central J 2008, Schlüter et al. Chem Central J 2009, J of Proteomics 134, Feb16, 2016, Special Issue “Towards deciphering proteomes via the proteoform, protein speciation, moonlighting and protein code concepts.